Руководство пользователя кластера OpenPOWER ВЦ ДВО РАН¶
- Руководство пользователя кластера OpenPOWER ВЦ ДВО РАН
Версия: 0.8
Дата: 09.04.2020
Обновлена инструкция по использованию пакетов ML/DL
Данное руководство содержит минимально необходимый объем информации для работы на гибридном кластере ВЦ ДВО РАН: описание процесса регистрации, сведения по работе в ОС Linux (вход в систему, работа с каталогами и файлами, мониторинг) и работе с MPI программами и непараллельными программами на кластере (компиляция, запуск, остановка, работа с очередями). В тексте под термином параллельная программа подразумеваются только MPI программы.
Команды и переменные командного интерпретатора, названия программ, листинги, непосредственный ввод/вывод консоли выделены моноширинным шрифтом.
Вопросы относительно работы кластера следует отправлять на e-mail: support@hpc.febras.net.
Вопросы относительно этого документа (ошибки, неточности, предложения) можно отправлять на e-mail: support@hpc.febras.net.
Описание системы¶
Гибридный вычислительный кластер состоит из одного управляющего и четырех вычислительных узлов. Все они представляют собой серверы Sitonica PW22LC (IBM Power Systems S822LC 8335-GTB). Каждый включает в себя два процессора IBM POWER8 с максимальной частотой 4,023 ГГц, два сопроцессора NVIDIA Tesla P100 GPU, 256 ГБ DDR4 ОЗУ, контроллер EDR InfiniBand и два жестких диска Seagate ST1000NX0313 1 ТБ 7200RPM. Для хранения пользовательских данных используется внешнее дисковое хранилищу объемом 50 ТБ. В качестве сети управления используется сеть, построенная по технологии Gigabit Ethernet. В качестве сети передачи данных используется сеть EDR InfiniBand с пропускной способностью в 100 Гбит/с.
Регистрация¶
Регистрация пользователей на кластере происходит через систему ЦКП http://ckp.ccfebras.ru.
Вход в систему¶
Для работы с системой пользователь должен иметь свою учетную запись на управляющем узле кластера. Регистрация пользователя на кластере происходит в соответствии с предыдущей частью руководства. После регистрации пользователь получает свое имя (логин), пароль и домашнюю директорию. Если имя пользователя, например, будет user, то домашняя папка находится в /home/user.
При первом входе в систему предлагается сменить пароль. Обратите внимание, что перед вводом нового пароля в строке (current) LDAP Password: необходимо ввести еще раз текущий пароль. Требования к новому паролю: он должен быть достаточной длины, содержать хотя бы 1 цифру и 1 заглавную букву. Директория ~/.ssh содержит пару ключей (id_rsa и authorized_keys) для доступа к узлам кластера. При добавлении личного ssh-ключа, необходимо добавить открытый ключ в файл authorized_keys через перенос строки (т.е. после уже имеющегося одного открытого ключа).
Пользователи имеют возможность работать на кластере с любой машины, находящейся в сети института и интернет. Для входа в систему пользователю необходим адрес сервера (http://jupiter.febras.net), а также имя и пароль, полученные при регистрации.
Вход с Windows-машины¶
Работа с системой осуществляется по безопасному протоколу SSH при помощи какого-либо ssh-клиента. Клиент должен поддерживать протокол версии 2. Рекомендуется использовать PuTTY. Эта программа является свободно распространяемой и проста в использовании.
После запуска программы пользователь должен выбрать протокол ssh и в поле «Host Name (or IP address)» указать адрес сервера. Нажатие на «Open» приведет к отправке запроса на подключение. В случае успешного подключения к серверу будет предложено ввести имя (логин), а затем и пароль.
Окно ssh-клиента PuTTY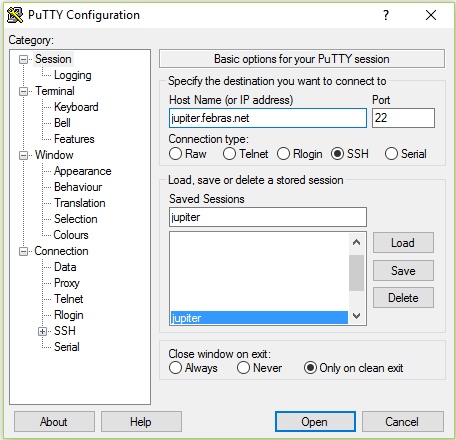
При вводе пароля символы на экране не отображаются. Если все введено правильно, то пользователь автоматически окажется в своей домашней директории. Этот каталог доступен пользователю с любого узла кластера.
Примечание. На кластере существует единое дисковое пространство для директорий /opt (только чтение) и /home. Все узлы используют дисковый массив сервера посредством сетевой файловой системы NFS. Файл записанный на одном из узлов кластера автоматически становится доступен на любом другом.
Работа в ssh-сессии происходит в терминальном (текстовом, консольном) режиме. Необходимо помнить, что консоль Linux, в отличии от Windows, различает регистр вводимых символов, то есть mydoc.txt и mydoc.TXT не одно и то же. После входа на экране отображается консоль командного интерпретатора в формате имя_пользователя@машина текущий_каталог:
[user@jupiter ~]$
Вход с терминала Linux¶
В любой дистрибутив ОС Linux входит терминальный ssh-клиент (обычно OpenSSH). Минимальный формат команды для подключения к кластеру таков:
[user@localhost ~]$ ssh jupiter.febras.net -l имя_пользователя
Копирование файлов¶
См. соответствующий раздел руководства кластера версии 5.0.
Навигация¶
См. соответствующий раздел руководства кластера версии 5.0.
Редактирование файлов¶
См. соответствующий раздел руководства кластера версии 5.0.
Environment Modules¶
На кластере используется система модулей окружения (Environment Modules). Загруженные модули можно посмотреть с помощью команды:
[user@jupiter ~]$ module list Currently Loaded Modulefiles: 1) core 2) xl/13.1-15.1 3) cuda 4) essl 5) spectrum_mpi
Указанные выше модули загружаются по умолчанию при входе в систему.
Список всех доступных модулей можно увидеть с помощью команды:
[user@jupiter ~]$ module avail ------------------------------------------------- /etc/modulefiles -------------------------------------------------- biobuilds/2017.11 mpi/gcc/2.0/4.8.5 pgi/17.4 conda/267/0.1 mpi/mvapich2-2.0-ppc64le pgi/18.4 conda/numba/0.39 mpi/mvapich2-ppc64le python/2.7 conda/pytorch/0.4.0 openmpi/gcc/1.10.6/4.8.5 python/3.5 core/1.0(default) openmpi/gcc/2.0.2a1/4.8.5 singularity/2.2.1 cuda/8.0.61(default) openmpi/gcc/2.1.0/4.8.5 singularity/2.3.1 cudnn/5.1 openmpi/pgi/1.10.2/16.10 singularity/2.4 espresso/cpu/5.4 openmpi/pgi/1.10.2/17.4 singularity/2.4.2 espresso/cpu/5.4-default-libs openmpi/xl/1.10.6/13.1-15.1 spectrum_mpi/gcc/10.1/4.8.5 espresso/gpu/5.4 openmpi/xl/2.0.2a1/13.1-15.1 spectrum_mpi/pgi/10.1 espresso/gpu/5.4-esslcuda openmpi/xl/2.1.0/13.1-15.1 spectrum_mpi/xl/10.1/13.1-15.1 espresso/gpu/6.0 openmpi/xl/3.0.0/13.1-15.1 spectrum-mpi/gcc/10.1/4.8.5 espresso/gpu/6.2-esslcuda pai/caffe/bvlc/1.0.0 spectrum-mpi/pgi/10.1 essl/5.5(default) pai/caffe/ibm/1.0.0 spectrum-mpi/xl/10.1/13.1-15.1 example/notavail pai/caffe/nvidia/0.15.14 tensorflow/1.1.0 gamess/07.10.2017-A pai/chainer/1.23.0 tensorflow/1.2.1 gamess/07.10.2017-B pai/digits/5.0.0 tensorflow/1.2.1-gpu gcc/4.8.5 pai/theano/0.9.0 utils/mpirun-pbswrap ibm-at/11.0 pai/torch/7 xl/13.1-15.1 modem/2016.05 pgi/16.10(default)
Компиляция программ¶
Доступные компиляторы¶
На кластере (на 29.08.2019) поддерживаются следующие компиляторы языков программирования для архитектуры ppc64le:
Компиляторы на кластере| Компилятор | Файл компилятора | Язык | Модуль |
|---|---|---|---|
| GNU C 4.8.5 | gcc | C | gcc/4.8.5 |
| GNU C++ 4.8.5 | g++ | C++ | gcc/4.8.5 |
| GNU Fortran 4.8.5 | gfortran | Fortran 90 | gcc/4.8.5 |
| GNU C 7.3.1 | gcc | C | ibm-at/11.0 |
| GNU C++ 7.3.1 | g++ | C++ | ibm-at/11.0 |
| GNU Fortran 7.3.1 | gfortran | Fortran 90 | ibm-at/11.0 |
| IBM XL Fortran 15.1.5 | xlf/xlf_r* | Fortran 77 | xl/13.1-15.1 |
| IBM XL Fortran 15.1.5 | xlf90/xlf90_r | Fortran 90 | xl/13.1-15.1 |
| IBM XL C 13.1.5 | xlc/xlc_r | C | xl/13.1-15.1 |
| IBM XL C++ 13.1.5 | xlc++/xlc++_r | C++ | xl/13.1-15.1 |
| PGI C 17.4 | pgcc | C | pgi/17.4 |
| PGI C++ 17.4 | pgc++ | C++ | pgi/17.4 |
| PGI Fortran 17.4 | pgf90 | Fortran | pgi/17.4 |
| PGI C 18.4 | pgcc | C | pgi/18.4 |
| PGI C++ 18.4 | pgc++ | C++ | pgi/18.4 |
| PGI Fortran 18.4 | pgf90 | Fortran | pgi/18.4 |
| PGI C 19.1 | pgcc | C | pgi/19.1 |
| PGI C++ 19.1 | pgc++ | C++ | pgi/19.1 |
| PGI Fortran 19.1 | pgf90 | Fortran | pgi/19.1 |
| PGI C 19.7 | pgcc | C | pgi/19.7 |
| PGI C++ 19.7 | pgc++ | C++ | pgi/19.7 |
| PGI Fortran 19.7 | pgf90 | Fortran | pgi/19.7 |
| NVIDIA Cuda 8.0.61 | nvcc | - | cuda/8.0.61 |
| NVIDIA Cuda 9.2.148 | nvcc | - | cuda/9.2.148 |
| NVIDIA Cuda 10.1.168 | nvcc | - | cuda/10.1.168 |
* - файлы компиляторов IBM с постфиксом "_r" используются для сборки безопасных с точки зрения потоков исполняемых файлов.
Пути к компиляторам GNU и IBM XL прописаны в переменной окружения «PATH» для всех пользователей. Дополнительно в переменной среды «LD_LIBRARY_PATH» указаны пути к библиотекам этих компиляторов. Поэтому загружать какие-либо модули для их использования не нужно. Для использования остальных компиляторов необходимо подгружать соответствующие модули.
Компиляция MPI программ¶
На кластере (на 16.02.2018) поддерживаются следующие реализации библиотеки MPI:
Библиотеки MPI на кластере| Библиотека MPI | Компилятор | Модуль |
|---|---|---|
| IBM Spectrum MPI 10.1 | GNU C/C++/Fortran 4.8.5 | spectrum-mpi/gcc/10.1/4.8.5 |
| IBM XL C/C++ 13.1.5 и Fortran 15.1.5 | spectrum-mpi/xl/10.1/13.1-15.1 | |
| PGI С/C++/Fortran 17.4 | spectrum-mpi/pgi/10.1 | |
| OpenMPI 2.0.2a1 | GNU C/C++/Fortran 4.8.5 | openmpi/gcc/2.0.2a1/4.8.5 |
| IBM XL C/C++ 13.1.5 и Fortran 15.1.5 | openmpi/xl/2.0.2a1 | |
| OpenMPI 1.10.2 | PGI С/C++/Fortran 17.4 | openmpi/pgi/1.10.2/17.4 |
По умолчанию для всех пользователей установлены переменные среды, позволяющие использовать библиотеку Spectrum MPI совместно с компиляторами IBM XL (модуль spectrum-mpi/xl/10.1/13.1-15.1). Для использования остальных реализаций MPI, или при необходимости использования других компиляторов, нужно загрузить соответствующий модуль. В связи с тем, что все реализации MPI конфликтуют между собой, перед загрузкой нового модуля необходимо выгружать текущий:
module unload xl/13.1-15.1 module unload spectrum-mpi/xl/10.1/13.1-15.1 module load gcc/4.8.5 module load openmpi/gcc/2.0.2a1/4.8.5
При использовании компилятора PGI требуется загрузить его модуль перед загрузкой модуля библиотеки MPI:
module unload xl/13.1-15.1 module unload spectrum-mpi/xl/10.1/13.1-15.1 module load pgi/17.4 module load openmpi/pgi/1.10.2/17.4
После подключения нужного модуля, пути к данной библиотеке добавляются в переменные среды «PATH» и «LD_LIBRARY_PATH».
Для компиляции mpi программ лучше всего использовать обёртки к компиляторам, чем вручную прописывать для этого специальные флаги. Так, например, чтобы скомпилировать mpi программу, написанную на языке Fortran, нужно воспользоваться оберткой mpifort. Данная команда вызовет компилятор IBM XL Fortran (при условии, что подключен модуль spectrum-mpi/xl/10.1/13.1-15.1), с указанием всех необходимых флагов.
Распишем соответствие между обертками и соответствующими им компиляторами:
Соответствие между обертками MPI и компиляторами| Обертка | XL | GNU | PGI |
|---|---|---|---|
| mpicc | xlc_r | gcc | pgcc |
| mpifort | xlf_r | gfortran | pgfortran |
| mpif77 | xlf_r | gfortran | pgfortran |
| mpif90 | xlf90_r | gfortran | pgfortran |
| mpic++ | xlC_r | g++ | pgc++ |
| mpicxx | xlC_r | g++ | pgc++ |
Для того, чтобы посмотреть какие опции компилятора указываются при вызове обертки, можно воспользоваться следующей командой:
[user@jupiter ~]$ mpicc --show gcc -I/opt/soft/openmpi/2.0.2a1/gcc/include -pthread -Wl,-rpath -Wl,/opt/soft/openmpi/2.0.2a1/gcc/lib -Wl, --enable-new-dtags -L/opt/soft/openmpi/2.0.2a1/gcc/lib -lmpi
Как видно из данного вывода, единственными опциями, которые может потребоваться указать при вызове компилятора, могут оказаться опции оптимизации.
Замечания по разработке программ на отдельной машине¶
Практически все реализации MPI поддерживают запуск параллельных приложений в режиме эмуляции на отдельно взятой рабочей станции. Это можно делать как на Linux, так и Windows машинах.
В Linux рекомендуется использовать пакет OpenMPI, а для создания MPI приложений на Windows машинах можно использовать пакет MPICH в версии для Windows. Для успешного портирования программ с Windows на Linux не следует использовать расширения предоставляемые средами программирования, такими как Visual Studio и Borland Builder.
Подготовленные исходные коды программ лучше всего компилировать на кластере.
Запуск задач¶
Диспетчеризация задач¶
Для диспетчеризации задач на кластере используется система PBS Professional 14.1. С её помощью пользователь может отправлять свои задачи на исполнение, снимать их с исполнения и получать информацию по текущему статусу задачи.
Данная система построена на основе очередей, где под очередью понимается набор пользовательских процессов (программ, задач) выполняющихся в рамках системы диспетчеризации. Каждой очереди сопоставлен ряд атрибутов, в зависимости от которых к задаче будут применены те или иные действия. Типичными атрибутами являются название (идентификатор) очереди, её приоритет, доступные ресурсы, количество задач. В общем случае термин очередь не означает, то что программы в ней будут выполняться строго последовательно.
Чтобы поставить задачу на исполнение, пользователь должен добавить ее при помощи команды qsub в какую-либо очередь. Очереди отличаются друг от друга совокупностью ресурсов, которыми они обладают.
Система очередей¶
На данный момент для пользователей действует единая очередь маршрутизации задач workq, в которой доступны все 5 узлов. Задачи из неё распределяются по очередям выполнения. Поставить задачу напрямую в очередь выполнения нельзя.
Для получения информации об очередях, можно выполнить команду qstat -q:
[eab@jupiter install]$ qstat -q
server: jupiter1
Queue Memory CPU Time Walltime Node Run Que Lm State
---------------- ------ -------- -------- ---- ----- ----- ---- -----
fastq -- -- 00:10:00 -- 0 0 -- E R
mixedq -- -- 390:00:0 -- 0 0 -- E R
mixedq-gpu -- -- -- -- 0 0 -- E R
workq -- -- -- -- 0 0 -- E R
lowq -- -- -- -- 0 0 -- E R
----- -----
0 0
Queue – имя очереди; Run – число выполняемых задач; Que – число задач, ожидающих начала выполнения. Команда qstat -Qf имя_очереди позволяет получить информацию о конкретной очереди.
Опция fastq¶
Сейчас отключена: fastq не привязана к
jupiter1[0]. Есть возможность подключить эту опцию в случае, если будет большая загруженность всех узлов и будет необходимость иметь всегда свободную небольшую часть ресурсов.
Задачи, для которых установлен walltime не более 10 минут, отправляются в короткую очередь fastq, привязанную к jupiter1[0] – первой половине узла jupiter1. Иначе говоря, часть ресурсов 1 узла кластера, в т.ч. и GPU0, доступна только для быстрых тестовых задач. Установить значение walltime можно с помощью строки:
#PBS -l walltime=0:10:00в файле задачи.
Задачу, которая запрашивает ресурсов больше, чем есть на 1 виртуальном узле, невозможно запустить с
walltime<=0:10:00, поскольку все такие задачи размещаются в очередь fastq, привязанную к виртуальному узлуjupiter1[0]. Поэтому, для любых больших задач необходимо задавать значениеwalltimeкак минимум0:10:01.
Задача сwalltime<=0:10:00обязательно попадает в очередь fastq, но не обязательно на узелjupiter1[0]: в случае, если есть другие свободные узлы, она может быть поставлена на один из них.
Постановка задачи в очередь¶
Для постановки задачи в очередь на исполнение используется команда qsub. Данная команда принимает в качестве параметра имя скрипта, в котором описываются требуемые задачей ресурсы и указываются команды, исполняемые при запуске. Рассмотрим пример, иллюстрирующий запуск ранее скомпилированной программы на 1 чанке (некоторой виртуальной части узла, которая не может по всем запрашиваемым ресурсам превышать размер одного узла) кластера, с использованием 4 mpi процессов, выделением 4 ядер и 1 GPU на этом чанке:
[user@jupiter mpi_test]$ cat mpi_test.qsub #PBS -k oe #PBS -l select=1:mpiprocs=4:ncpus=32:mem=4gb:ngpus=1 #PBS -l walltime=1:00:00 #PBS -l place=shared #PBS -r n #PBS -M user@mail.com #PBS -m abe #PBS -q workq #PBS -N mpi_test #!/bin/sh cd /home/user/test/mpi_test module unload xl/13.1-15.1 && module load gcc/4.8.5 module unload spectrum_mpi && module load openmpi/gcc/1.10.6/4.8.5 mpirun -np 4 -npernode 4 --hostfile $PBS_NODEFILE --bind-to core ./mpi [user@jupiter mpi_test]$ qsub mpi_test.qsub 66330.jupiter1
Если команда выполнена успешно, то на экране отобразится идентификатор задачи (в данном случае это 66330.jupiter1), в противном случае появится сообщение об ошибке. Ошибки пользовательской программы (неправильная компиляция и т.п.) проявятся только при переходе задачи к активному состоянию.
Примечание. Весь вывод программы в стандартный поток и в поток ошибок перенаправляется в файлы, находящиеся в домашней директории пользователя. Названия таких файлов имеют формат
имя_задачи.(e/o)порядковый_номер. Для запущенной задачи это будут: mpi_test.e66330 – для потока ошибок и mpi_test.o66330 – для стандартного потока вывода.
Прокомментируем каждую из строчек скрипта mpi_test.qsub:
#PBS -k oe — указание сброса потока вывода (o) и потока ошибок (e)#PBS -l select=1:mpiprocs=4:ncpus=32:mem=4gb:ngpus=1 — требуемое количество чанков (1); количество mpi процессов (4), количество выделяемых ядер (4 = 32/8), количество оперативной памяти и количество выделяемых GPU (1) на каждом чанке
Параметр ngpus=<число> является обязательным к указанию для задач, планируемых к запуску с использованием графических ускорителей. <число> — количество ускорителей, требуемое задачей.
#PBS -l walltime=1:45:00 — максимальное время исполнения задачи (в данном случае - 1 час 45 минут), после истечения которого она будет снята планировщиком#PBS -l place=shared — использование узла вместе с другими задачами; shared — совместное использование, excl — монопольное использование#PBS -r n — является ли задача перезапускаемой (задачей с контрольными точками); y — является, n — не является#PBS -M user@mail.com — почтовый адрес пользователя#PBS -m abe — какие сообщения отправляются на указанный адрес (a — ошибка в выполнении задачи, b — начало выполнения, e — завершение выполнения)#PBS -q workq — идентификатор очереди#PBS -N mpi_test — название задачи#!/bin/sh — указание необходимого командного интерпретатораcd /home/user/test/mpi_test — переход в директорию с исполняемым файломmodule unload xl/13.1-15.1 && module load gcc/4.8.5 — выбор gcc вместо xlmodule unload spectrum_mpi && module load openmpi/gcc/1.10.6/4.8.5* — выбор openmpi вместо spectrum_mpimpirun -np 4 -npernode 4 --hostfile $PBS_NODEFILE --bind-to core ./mpi — запуск приложения
Запуск интерактивных программ¶
Запуск управляющей консоли через планировщик¶
Вы можете запустить интерактивную задачу, используя директиву qsub -I. Например:
qsub -I -N jobname -l select=1:ncpus=8:mem=1gb:ngpus=1,walltime=1:00:00После запуска установленная по умолчанию командная оболочка, например bash, откроется на узле, выделенном планировщиком для выполнения задания.
Вы не можете использовать PBS скрипт для запуска интерактивной задачи.
Запуск программ с фиксированным вводом¶
Программы, использующие стандартный ввод, называются интерактивными. Как правило, такие программы после запуска требуют от пользователя ввода данных. При постановке задачи в очередь любая программа переводится в фоновый режим. В этом режиме ввод данных пользователем в запущенную программу невозможен. Для передачи данных таким программам используется механизм перенаправления стандартных потоков ввода/вывода.
Для перенаправления подготавливается текстовый файл, содержимое которого в точности представляет собой данные, вводимые пользователем. Например, если программа solver предполагает ввод в первой строке размерности матрицы, а во второй количества итераций, то текстовый файл input.txt будет иметь вид:
[user@jupiter solver]$ cat input.txt 10000000 1000
После каждого числа обязателен символ новой строки. Запуск программы на выполнение производится так:
solver < input.txt
Скрипт для постановки в очередь задания, в рамках которого будет выполняться интерактивная программа, будет выглядеть следующим образом:
[user@jupiter solver]$ cat job.qsub #PBS -k oe #PBS -l select=1:ncpus=64:mem=8gb:mpiprocs=8 #PBS -r n #PBS -M user@mail.com #PBS -m abe #PBS -q workq #PBS -N solver #!/bin/sh cd /home/user/test/solver mpirun -np 8 -npernode 8 --hostfile $PBS_NODEFILE --bind-to core ./solver < ./input.txt exit 0
Запуск непараллельных программ¶
Запуск непараллельных программ практически ничем не отличается от запуска параллельных программ. Единственное отличие заключается в том, что в qsub скрипте такой программы необходимо указать, что для её работы необходимо только одно ядро:
#PBS -l select=1:ncpus=8:mem=1gb
Также в этом скрипте необходимо запускать непосредственно исполняемый файл программы, то есть не использовать для запуска mpirun.
Состояние пользовательских задач¶
Для получения информации об очередях и задачах пользователя используется команда qstat. Выполнение этой команды без параметров покажет все задачи пользователя и их состояние.
[user@jupiter ~]$ qstat Job id Name User Time Use S Queue ----------------------- ---------------- --------------- -------- - ----- 700.jupiter1 mpi_test user 00:10:40 R workq 701.jupiter1 sample_job user 0 Q workq 702.jupiter1 solver user 0 Q workq
Job id — идентификатор задачи, полученный при выполнении qsub; Name — имя задачи; User — имя пользователя, запустившего задачу; Time Use — процессорное время, потраченное задачей; S (State) — состояние задачи ( R – задача выполняется, Q – ожидает в очереди); Queue — очередь.
В данном случае пользователю user принадлежат три задачи.
С помощью команды qstat -n идентификатор_задачи можно получить список узлов, на которых выполняется конкретная задача. Эта информация полезна при мониторинге эффективности использования вычислительных ресурсов с использованием системы Ganglia, так как позволяет отслеживать состояние только используемых задачей узлов.
[user@jupiter ~]$ qstat -n 701
jupiter1:
Req'd Req'd Elap
Job ID Username Queue Jobname SessID NDS TSK Memory Time S Time
--------------- -------- -------- ---------- ------ --- --- ------ ----- - -----
298.jupiter1 eab workq mpi_test -- 1 4 2gb 00:00 R --
jupiter1/0*4
Для получения более подробной информации о конкретной задаче можно запустить команду qstat -f идентификатор_задачи. Для просмотра информации по завершенным задачам можно использовать команды qstat -x | tail и qstat -xf идентификатор_задачи.
Остановка задач¶
Остановка программы производится командой qdel идентификатор_задачи
[user@jupiter ~]$ qdel 700
Этой командой задача, стоящая в очереди, убирается из нее, а выполняющаяся задача снимается с выполнения. Следующая по очереди и приоритету задача встает на выполнение.
Задача снимается в течении некоторого времени, поэтому при вызове qstat непосредственно после qdel удаленная задача все еще может быть отражена в таблице.
Углубленная настройка задач¶
Каждый узел кластера состоит из двух виртуальных узлов, разделяющих ресурсы узла поровну. Для оперативной памяти действует исключение: она может заимствоваться из соседнего виртуального узла.
Соответствие физических и программных ресурсов¶
Соответствие между физическими ресурсами узлов и программными ресурсами, которыми оперирует планировщик, не всегда прозрачно и очевидно. Попробуем разобраться в этом.
Физический узел кластера содержит 2 процессора, что соответствует 20 вычислительным ядрам или 160 потокам выполнения. В планировщике же задается такая конфигурация, что узел состоит из двух виртуальных узлов, на каждом из которых есть 80 вычислительных процессоров, обозначаемых как ncpus (т.е. вычислительный процессор планировщика соответствует потоку выполнения физического устройства). В зависимости от ситуации словом процессор обозначаются разные сущности. Чтобы прояснить ситуацию, рассмотрим гипотетический вариант конфигурации планировщика, когда виртуальный узел состоит из ncpus=10 вычислительных процессоров (т.е. вычислительный процессор планировщика соответствует ядру физического устройства). Задача, запускаемая планировщиком, не может не потреблять вычислительные ресурсы, то есть минимальное значение для ncpus задачи есть 1. Таким образом, в текущей конфигурации на узле можно одновременно запустить максимум 160 задач, а в конфигурации, приведенной для сравнения, только 20.
Мы неоднократно упоминали о рекомендации использовать значение ncpus примерно в 8 раз большее требуемого количества ядер. В этом случае, если минимальное значение ncpus равно 8 (что соответствует одному ядру), то максимальное количество задач, которые одновременно можно запустить на одном узле, — те же 20, что и во второй конфигурации. В чем же тогда отличие и преимущество конфигурации с ncpus=160? Её очевидным недостатком является необходимость понимать и помнить, что 8 ncpus это "на самом деле" 1 ядро.
Использование гиперпоточности (в конфигурации с ncpus=160 у каждого вычислительного ядра есть 8 потоков) в общем случае позволяет улучшить общую пропускную способность вычислительной системы (в среднем выполнять большее количество задач за данное время), потенциально уменьшает плату за единицу потребляемых ресурсов, а также в некоторых случаях, позволяет задаче, ожидающей в очереди, запуститься раньше. В то же время, использование гиперпоточности в некоторых случаях мешает достижению наилучшей производительности вычислительной системы.
Продемонстрируем это на примере. Предположим, есть задача, которой оптимально требуется 20 ядер CPU и не требуется GPU. Также, есть полностью свободный узел кластера:
mem ncpus nmics ngpus
vnode state njobs run susp f/t f/t f/t f/t jobs
jupiter3[0] free 0 0 0 126gb/126gb 80/80 0/0 1/1 --
jupiter3[1] free 0 0 0 126gb/126gb 80/80 0/0 1/1 --
Следующий запрос ресурсов
#PBS -l select=2:mpiprocs=10:ncpus=72:mem=4gb #PBS -l place=packпозволяет такой задаче разместиться (здесь параметр
place=pack обязует планировщика использовать строго один физический узел для чанков из запроса) на обоих виртуальных узлах
mem ncpus nmics ngpus
vnode state njobs run susp f/t f/t f/t f/t jobs
jupiter3[0] free 1 1 0 122gb/126gb 8/80 0/0 1/1 1083
jupiter3[1] free 1 1 0 122gb/126gb 8/80 0/0 1/1 1083
при этом оставляя свободные ncpus на каждом из них.
Если указаны правильные параметры
$MPIEXEC --bind-to core a.outдля mpi и
export OMP_PLACES=coresдля OpenMP, то задача равномерно и оптимально разместится на имеющихся 20 физических ядрах (используя только 20 потоков выполнения из 160), а планировщик запомнит, что задача заняла "почти все" (144 из 160) ресурсы CPU на этом узле.
Другая задача с запросом
#PBS -l select=1:ncpus=8:mem=40gb:ngpus=1может занять любой из все еще свободных виртуальных узлов
mem ncpus nmics ngpus
vnode state njobs run susp f/t f/t f/t f/t jobs
jupiter3[0] job-busy 2 2 0 82gb/126gb 0/80 0/0 0/1 1083,1084
jupiter3[1] free 1 1 0 122gb/126gb 8/80 0/0 1/1 1083
задействуя одно физическое ядро (используя еще 1 поток из оставшихся 140) и 1 GPU. Теперь виртуальный узел jupiter3[0] имеет состояние job-busy и не может больше принимать задачи.
Каков результат такого размещения задач с точки зрения эффективности использования ресурсов? Одно ядро системы вынуждено обслуживать 2 задачи (1083 и 1084), а оставшиеся 19 ядер оптимально работают с одной задачей (1083). Предположим,в благоприятном случае, балансировщик нагрузки ядра linux, за счет имеющегося в наличии большого количества свободных потоков, позволяет добиться того, что потери в итоговом времени выполнения задач 1083 (которой оптимально нужно 20 ядер) и 1084 (которой оптимально нужно 1 ядро) будут сведены к минимуму. Проще говоря, они практически не помещают друг другу. Другой вероятный сценарий: из-за того, что на каком-то ядре висит 2 задачи, они будут вычисляться, например, в 1.5 раза дольше, чем если бы они использовали ресурсы эксклюзивно. Если при этом еще и параллелизм задачи 1083 устроен так, что происходят регулярные синхронизации, когда все потоки должны завершиться, ожидая самый медленный, то перегрузка одного ядра сильно отразится на производительности для задачи 1083 в целом.
Определение количества mpi-процессов¶
На каждом узле кластера имеется 2 десятиядерных процессора, каждое ядро может обрабатывать 8 потоков. В скриптах запуска для параметра ncpus максимальным является значение 160 — общее количество потоков. Тем не менее, при определенных условиях (если код запускаемых программ хорошо оптимизирован), количество вычислительных процессов значительно большее, чем 20 доступных ядер, может стать причиной неэффективного выполнения — задачи будут мешать друг другу. Поэтому, если нет особых оснований, рекомендуется использовать 20 mpi-процессов на узле. Таким образом, количество запускаемых mpi-процессов должно примерно соответствовать количеству запрашиваемых ncpus, поделенному на 8. В некоторых случаях допускаются отклонения от этой схемы, например, для приложения GAMESS запускается в 2 раза больше потоков, чем обычно, и половина из них не требует отдельного процессора. В данном случае количество mpi-процессов будет соответствовать количеству ncpus, поделенному на 4.
В силу специфики узла кластера, имеющего 2 GPU, для задач, использующих все 20 mpi-процессов и не использующих GPU, рекомендуется устанавливать значение select=2:ncpus=72:mem=100gb (при этом обязательно указать place=pack), чтобы было возможно размещение еще 2х задач, не требовательных к CPU и задействующих GPU, с параметрами ncpus=8:ngpus=1:mem=25gb.
Использование MPS-сервера¶
На данный момент на кластере можно использовать CUDA MPS сервер. Настройка пока доступна только для задач, требующих 1 GPU и помещающихся ровно на 1 виртуальный узел (что означает задание ncpus<=80 при полностью свободных процессорных ресурсах виртуального узла). Чтобы активировать этот режим, необходимо в скрипте запуска указать:
#PBS -v USE_MPS=True
MPS-сервер будет создан при запуске задачи и удален после её завершения.
Согласование параметров mpirun¶
До настоящего времени приходилось дублировать параметры, передаваемые в запрос ресурсов, и параметры mpirun, например:
#PBS -l select=2:ncpus=144:mpiprocs=20:mem=200gb mpirun -np 40 --hostfile $PBS_NODEFILE --bind-to core a.outЗдесь количество mpi-процессов из запроса (2 чанка по 20 процессов) необходимо повторять в параметре
-np. Кроме того, необходимо передавать параметр --hostfile.
Теперь, с помощью экспериментального режима, запускаемого с помощью модуля utils/mpirun-pbswrap, можно заменять вызов mpirun на вызов обертки, которая позволяет избавиться от такого дублирования:
#PBS -l select=2:ncpus=144:mpiprocs=20:mem=200gb module load utils/mpirun-pbswrap mpirun --bind-to core a.out
Обертка поддерживается для всех модулей spectrum mpi и openmpi/gcc/2.1.0/4.8.5.
Мониторинг¶
Web-интерфейс¶
Мониторинг кластера реализован при помощи системы Ganglia. Эта система позволяет следить за ресурсами кластера посредством web-интерфейса. Система мониторинга находится по адресу http://jupiter.febras.net/ganglia.
Для мониторинга пользователю доступно большое число типов ресурсов: загруженность процессора, оперативная память, загрузка сети, средняя загрузка, количество процессов и ряд других. Имеется возможность наблюдать как за всеми узлами в кластере (по одному параметру), так и за каждым (по всем параметрам).
Консоль¶
Кроме графического интерфейса существует несколько полезных консольных команд для мониторинга. Команда pbsnodes имя_узла позволяет получить информацию о конкретном узле: тип, состояние, количество процессоров, выполняющиеся задачи. Ниже представлен фрагмент вывода этой команды.
[user@jupiter ~]$ pbsnodes jupiter2
jupiter2
Mom = jupiter2
Port = 15002
pbs_version = 14.1.0
ntype = PBS
state = free
pcpus = 160
resources_available.arch = linux
resources_available.host = jupiter2
resources_available.mem = 263653568kb
resources_available.ncpus = 160
resources_available.ngpus = 2
resources_available.vnode = jupiter2
resources_assigned.accelerator_memory = 0kb
resources_assigned.mem = 0kb
resources_assigned.naccelerators = 0
resources_assigned.ncpus = 0
resources_assigned.netwins = 0
resources_assigned.ngpus = 0
resources_assigned.vmem = 0kb
resv_enable = True
sharing = default_shared
state – состояние узла (job-exclusive – все ресурсы узла заняты; free – на узле есть свободные ресурсы для запуска заданий; offline – узел временно выведен из эксплуатации, запуск заданий на нем невозможен; down – узел выключен); pcpus – число процессорных потоков на узле; jobs – задачи, запущенные на узле; resources_available.ngpus – число GPU на узле; resources_available.nodetype — тип узла: jupiter (CentOS).
При выполнении команды pbsnodes -a -S -j будет выведена сводная информация обо всех узлах кластера.
[user@jupiter ~]$ pbsnodes -a -S -j
mem ncpus nmics ngpus
vnode state njobs run susp f/t f/t f/t f/t jobs
--------------- --------------- ------ ----- ------ ------------ ------- ------- ------- -------
jupiter1 free 0 0 0 251gb/251gb 160/160 0/0 2/2 --
jupiter2 free 0 0 0 251gb/251gb 160/160 0/0 2/2 --
jupiter3 free 0 0 0 251gb/251gb 160/160 0/0 2/2 --
jupiter4 free 0 0 0 251gb/251gb 160/160 0/0 2/2 --
jupiter5 free 0 0 0 251gb/251gb 160/160 0/0 2/2 --
При выполнении команды pbsnodes -v -a -S -j сводная информация будет выведена с внутренней детализацией.
[user@jupiter ~]$ pbsnodes -v -a -S -j
mem ncpus nmics ngpus
vnode state njobs run susp f/t f/t f/t f/t jobs
--------------- --------------- ------ ----- ------ ------------ ------- ------- ------- -------
jupiter2 free 0 0 0 0 b/0 b 0/0 0/0 0/0 --
jupiter3 free 0 0 0 0 b/0 b 0/0 0/0 0/0 --
jupiter3[0] free 2 2 0 126gb/126gb 52/80 0/0 0/1 5272,5326
jupiter3[1] free 1 1 0 126gb/126gb 72/80 0/0 0/1 5273
jupiter1 free 0 0 0 0 b/0 b 0/0 0/0 0/0 --
jupiter4 free 0 0 0 0 b/0 b 0/0 0/0 0/0 --
jupiter5 free 0 0 0 0 b/0 b 0/0 0/0 0/0 --
jupiter4[0] free 1 1 0 126gb/126gb 72/80 0/0 0/1 5278
jupiter4[1] free 1 1 0 126gb/126gb 72/80 0/0 0/1 5279
jupiter5[0] free 1 1 0 126gb/126gb 72/80 0/0 0/1 5280
jupiter5[1] free 1 1 0 126gb/126gb 72/80 0/0 0/1 5281
jupiter1[0] free 1 1 0 126gb/126gb 72/80 0/0 0/1 5276
jupiter1[1] free 1 1 0 126gb/126gb 72/80 0/0 0/1 5277
jupiter2[0] free 1 1 0 126gb/126gb 72/80 0/0 0/1 5275
jupiter2[1] free 1 1 0 126gb/126gb 72/80 0/0 0/1 5274
Пакеты прикладных программ¶
Фреймворки машинного и глубокого обучения¶
Менеджер окружений Anaconda¶
Для работы с Python-библиотеками в области Data Science и Machine Learning удобным инструментом является дистрибутив Anaconda. Он включает версию интерпретатора Python c набором необходимых базовых библиотек, а также manager зависимостей conda, позволяющий устанавливать необходимые пакеты. Кроме того, он позволяет создавать независимые изолированные окружения, содержащие свою версию Python и набор установленных пакетов.
На кластере Anaconda установлена в директорию /opt/soft/anaconda3, включить ее использование можно командой:
user@jupiter# source /opt/soft/anaconda3/bin/activate
Будет активировано базовое окружение по-умолчанию. Далее, создается новое окружение:
user@jupiter# conda create --name <envname>
В нем ставятся необходимые пакеты:
conda install <package>
Для более детальной информации по менеджеру conda следует ознакомиться с руководством#
PowerAI¶
На кластере доступен IBM Watson Machine Learning Community Edition (ранее - PowerAI) - набор пакетов, включающий оптимизированные для архитектуры POWER версии популярных ML/DL фреймворков и инструментов.
| Название | Версия | Доп. информация |
|---|---|---|
| IBM-enhanced Caffe | 1.0.0 | Фреймоворк для глубокого обучения |
| Keras | 2.3.1 | Библиобека для построения нейронных сетей |
| PyTorch | 1.3.1 | Библиотека машинного обучения для языка Python, созданная на базе Torch - набор алгоритмов глубокого обучения и научных расчётов |
| TensorFlow | 2.1.0 | Библиотека для машинного обучение, разработанная Google |
| DDL | 1.5.1 | Библиотека от IBM, обеспечивающая распределенное выполнения для TensorFlow, Caffe, Torch |
| Horovod | 0.19 | Библиотека, использующая NCCL и MPI для распределенного запуска TensorFlow |
| NVIDIA NCCL | 2.5.6 | Библиотека, предоставляющая базовые примитивы для взаимодействия множества GPU на разных узлах |
| NVIDIA cuDNN | 7.6.5 | Библиотека для ускорения работы глубоких нейронных сетей на NVIDIA GPU |
| NVIDIA DALI | 0.18 | Библиотека, ускоряющая препроцессинг входных данных для нейронных сетей |
| NVIDIA TensorRT | 7.0.0.11 | Высокопроизводительная библиотека для инференса нейронных сетей |
| RAPIDS | 0.11.0 | |
| XGBoost | 0.90 | Высокопроизводительная реализация Gradient Boosting для Python |
PowerAI, необходимо установить и активировать окружение с помощью следующих команд (в качестве имени окружения для примера будет использоваться powerai, на деле оно может быть любым, кроме имен уже существующих окружений).
- Загрузить базовое окружение Anaconda:
user@jupiter# source /opt/soft/anaconda3/bin/activate
После этого станут доступны окружения PowerAI, установленные на кластере:user@jupiter# conda env list # conda environments: # base * /opt/soft/anaconda3 wmlce-1.6.2-py36 /opt/soft/anaconda3/envs/wmlce-1.6.2-py36 wmlce-1.6.2-py37 /opt/soft/anaconda3/envs/wmlce-1.6.2-py37 wmlce-1.7.0-py36 /opt/soft/anaconda3/envs/wmlce-1.7.0-py36 wmlce-1.7.0-py37 /opt/soft/anaconda3/envs/wmlce-1.7.0-py37
Можно активировать необходимое, и работать с ним, если не требуется установка каких-либо других пакетов, не входящих в окружение (можно посмотреть командойconda list). - Если требуется установка недостающих пакетов, то окружение необходимо склонировать в домашнюю директорию и активировать. Пример команды:
user@jupiter# conda create --name powerai --clone wmlce-1.7.0-py36 user@jupiter# conda activate powerai
Установка дополнительных пакетов:user@jupiter# conda install <package>
- При клонировании уже установленного на кластере powerai-окружения будут скопированы все входящие в его состав фреймворки и инструменты (~15Гб). Если требуется работа с одним или несколькими конкретными пакетами, можно настроить окружение с нуля вручную.
user@jupiter# export IBM_POWERAI_LICENSE_ACCEPT=yes user@jupiter# conda config --prepend channels file:///install/soft/ibm/WMLCE/repo user@jupiter# conda config --set channel_priority strict user@jupiter# conda create --name powerai python=3.6 powerai-release=1.7.0 user@jupiter# conda activate powerai user@jupiter# conda install --copy cudatoolkit-dev python=3.6 powerai-release=1.7.0
Установка требуемого фреймворка:user@jupiter# conda install --copy <package> python=3.6 powerai-release=1.7.0
Возможные значения <package>: caffepytorchtensorflow-gpuhorovodkeras-gpusnapml-sparkpy-xgboost-gpupowerai-rapidsmagmabazel
С полным перечнем и описаниями пакетов можно ознакомиться на сайте
Для запуска задач на расчет используется следующий код в qsub-скрипте:
# отключаем версию CUDA по-умолчанию, требуемая версия поставляется вместе с PowerAI module unload cuda/8.0.61 # включаем Anaconda source /opt/soft/anaconda3/bin/activate # активируем окружение conda activate powerai # обеспечиваем использование нужной версии драйвера export LD_LIBRARY_PATH=$CONDA_PREFIX/compat:$LD_LIBRARY_PATH # Переходим в рабочую директорию проекта cd /home/<username>/tf_working_dir/cifar10/ # Запускаем тренировку нейронной сети, описанной в файлах cifar10_multi_gpu_train.py python cifar10_multi_gpu_train.py
Использование нескольких GPU¶
Использование нескольких GPU позволяет ускорить расчет градиентов обновления весов сети. Для этого несколько копий модели параллельно просчитываются на отдельных GPU, после чего вычисляется средний градиент ошибки для обновления весов сети.
Ниже приведены части кода, исполняющего задачу классификации изображений из набора CIFAR-10, с помощью сверточной нейронной сети. Полный код приведен в файлах:
cifar10_multi_gpu_train.py – задает процесс обучения на нескольких графических сопроцессорах;cifar10.py – содержит вспомогательные функции, а также описывает вычислительный граф;cifar10_input.py – загружает и структурирует данные из набора CIFAR-10;cifar10_eval.py – загружает обученную модель и оценивает точность тренировки нейронной сети.
Для параллелизации процесса обучения в файле cifar10_multi_gpu_train.py задается параметр FLAGS.num_gpus, представляющий собой количество GPU (задается в tf.app.flags.DEFINE_integer).
Копии модели в TensorFlow принято называть tower. Ниже приведен пример, в котором каждая tower рассчитывается на отдельном GPU, после чего вычисляется средний градиент весов сети:
tower_grads = []
with tf.variable_scope(tf.get_variable_scope()):
for i in xrange(FLAGS.num_gpus):
with tf.device('/gpu:%d' % i):
with tf.name_scope('%s_%d' % (cifar10.TOWER_NAME, i)) as scope:
# Dequeues one batch for the GPU
image_batch, label_batch = batch_queue.dequeue()
# Calculate the loss for one tower of the CIFAR model. This function
# constructs the entire CIFAR model but shares the variables across
# all towers.
loss = tower_loss(scope, image_batch, label_batch)
# Reuse variables for the next tower.
tf.get_variable_scope().reuse_variables()
# Retain the summaries from the final tower.
summaries = tf.get_collection(tf.GraphKeys.SUMMARIES, scope)
# Calculate the gradients for the batch of data on this CIFAR tower.
grads = opt.compute_gradients(loss)
# Keep track of the gradients across all towers.
tower_grads.append(grads)
# We must calculate the mean of each gradient. Note that this is the
# synchronization point across all towers.
grads = average_gradients(tower_grads)
Хранение данных на кластере¶
При работе с фреймворком TensorFlow настоятельно рекомендуется хэшировать данные в директории /tmp, имеющейся на каждом вычислительном узле. Эти директории находятся на локальных дисках объемом 2Tb. Для исключения конфликтов между данными разных пользователей на одном узле, необходимо размещать их в директории /tmp/username, как это сделано в коде ниже.
username = str(os.environ['USER'])
tf.app.flags.DEFINE_string('data_dir', '/tmp/'+username+'/cifar10_data', """Path to the CIFAR-10 data directory.""")
Полное руководство по созданию данной модели глубокой нейронной сети в TensorFlow приведено по ссылке: https://www.tensorflow.org/tutorials/deep_cnn
Quantum ESPRESSO¶
Quantum ESPRESSO — это интегрированный набор машинных кодов для энергетических расчетов многоэлектронных систем. Программа основана на теории функциональной плотности, плоских волн и псевдопотенциалов. Свободное программное обеспечение, доступное для всех пользователей кластера.
Версии¶
Версии пакета| Версия | Используемая библиотека | Поддержка GPU | Модуль | Сайт проекта |
|---|---|---|---|---|
| 5.4 | NETLIB, FFTW | - | espresso/cpu/5.4-default-libs | http://www.quantum-espresso.org/ |
| 5.4 | IBM ESSL | - | espresso/cpu/5.4 | http://www.quantum-espresso.org/ |
| 5.4 | IBM ESSL | +* | espresso/gpu/5.4 | https://github.com/fspiga/qe-gpu-plugin |
| 5.4 | IBM ESSL (SMPCUDA) | ++ | espresso/gpu/5.4-esslcuda | http://www.quantum-espresso.org/ |
| 6.0 | IBM ESSL | +++ | espresso/gpu/6.0 | https://github.com/RSE-Cambridge/qe-gpu |
* – число символов “+” соотносится с производительностью пакета (больше – лучше)
Все версии пакета собраны с поддержкой MPI и могут работать в параллельном режиме. Версия Quantum ESPRESSO 6.0 является экспериментальной и может не поддерживать некоторые функции и настройки (например, в этой версии нельзя выставить значение nk большее 1; подробности смотрите на сайте проекта).
Запуск Quantum ESPRESSO¶
Перед началом работы с пакетом необходимо загрузить соответствующий модуль командой “module add”. Это позволит получить доступ к нужной версии исполняемых файлов пакета и выставить необходимые переменные среды. Также может потребоваться загрузить дополнительные модули, от которых зависит работоспособность соответствующей версии Quantum ESPRESSO. Все эти действия выполняются в скриптах описания заданий.
Ниже приводятся примеры скриптов описания заданий, используемых для запуска Quantum ESPRESSO в параллельном режиме на узлах кластера (два вычислительных узла; используются 2 процессора, т.е. 20 ядер, и 2 сопроцессора на каждом).
Версия Quantum Espresso – espresso/cpu/5.4 и espresso/cpu/5.4-default-libs.
#!/bin/sh #PBS -j oe #PBS -l select=2:ncpus=144:mpiprocs=20:mem=200gb #PBS -l walltime=0:48:00 #PBS -q workq #PBS -N qe_job_name # Загружаем необходимые модули # При использовании espresso/cpu/5.4-default-libs загружаем # данный модуль вместо espresso/cpu/5.4 module add spectrum_mpi essl espresso/cpu/5.4 # Переходим в рабочую директорию cd /home/user/qe_working_dir # Запускаем исполняемый файл Quantum ESPRESSO “pw.x” mpirun -np 40 -npernode 20 --hostfile $PBS_NODEFILE --bind-to core $QE_HOME/bin/pw.x < ausurf.in
Примечание. Здесь запрос с
ncpus=144практически эквивалентен запросу сncpus=160с той разницей, что остается возможность для другой, не требовательной к cpu, небольшой задачи сncpus=8:ngpus=1занять свободные ресурсы на узле.
Версия Quantum ESPRESSO – espresso/gpu/5.4.
#!/bin/sh #PBS -j oe #PBS -l select=2:ncpus=160:ngpus=2:mpiprocs=20:mem=251gb #PBS -l walltime=48:00:00 #PBS -q workq #PBS -N qe_job_name # Загружаем необходимые модули module add spectrum_mpi essl espresso/gpu/5.4 # Переходим в рабочую директорию cd /home/user/qe_working_dir # Запускаем исполняемый файл Quantum ESPRESSO “pw-gpu.x” mpirun -np 40 -npernode 20 --hostfile $PBS_NODEFILE --bind-to core $QE_HOME/bin/pw-gpu.x < ausurf.in
Версия Quantum ESPRESSO – espresso/gpu/5.4-esslcuda.
#!/bin/sh #PBS -j oe #PBS -l select=2:ncpus=160:ngpus=2:mpiprocs=20:mem=251gb #PBS -l walltime=48:00:00 #PBS -q workq #PBS -N qe_job_name # Загружаем необходимые модули module add spectrum_mpi essl espresso/gpu/5.4-esslcuda # Переходим в рабочую директорию cd /home/user/qe_working_dir # Запускаем исполняемый файл Quantum ESPRESSO “pw.x” mpirun -np 40 -npernode 20 --hostfile $PBS_NODEFILE --bind-to core $QE_HOME/bin/pw.x < ausurf.in
Версия Quantum ESPRESSO – espresso/gpu/6.0.
#!/bin/sh #PBS -j oe #PBS -l select=2:ncpus=160:ngpus=2:mpiprocs=2:mem=251gb #PBS -l walltime=48:00:00 #PBS -q workq #PBS -N qe_job_name # Загружаем необходимые модули module rm spectrum_mpi xl/13.1-15.1 module add pgi/17.4 spectrum-mpi/pgi/10.1 essl espresso/gpu/6.0 # Переходим в рабочую директорию cd /home/user/qe_working_dir # Запускаем исполняемый файл Quantum ESPRESSO “pw.x” mpirun -np 4 --hostfile $PBS_NODEFILE --map-by ppr:1:socket $QE_HOME/bin/pw.x < ausurf.in
Стандартный вывод и вывод ошибок перенаправляется в файл вида qe_job_name.o1079, где “1079” соответствует номеру задания.
GAMESS¶
GAMESS — программа пакета вычислительной квантовой химии. GAMESS (US) способен производить расчеты МО при помощи многих методов вычислительной квантовой химии, включая метод Хартри-Фока, теорию функционала плотности (DFT), теорию валентных связей (GVB), и многоконфигурационное самосогласованное поле (MCSCF).
Версии¶
Установлена и протестирована версия GAMESS 2017 R2 с использованием ESSL и OpenMPI 2.0.2a1.
Для версии GAMESS+LIBCCHEM с использованием вычислений на GPU, компиляция проходит успешно, но часть тестов связанных с вычислением MP2 energy проваливаются.
Запуск GAMESS¶
Ниже приводится пример скрипта описания задания, используемого для запуска GAMESS в параллельном режиме на узлах кластера (два вычислительных узла).
#!/bin/sh #PBS -N gm_job_name #PBS -q workq #PBS -k oe #PBS -l select=2:ncpus=144:mpiprocs=40:mem=200gb #PBS -l walltime=48:00:00 #PBS -m abe # Загружаем необходимые модули module rm xl/13.1-15.1 module rm spectrum_mpi module add gcc/4.8.5 module add openmpi/gcc/2.0.2a1/4.8.5 module add gamess/07.10.2017-A # Переходим в рабочую директорию cd /home/user/gm_working_dir # Запуск GAMESS с входным файлом SubPc.inp. # rungms принимает 4 параметра: # имя входного файла (SubPc.inp) # версия GAMESS (01) # количество ядер NCPUS (40) # количество ядер на узел PPN (20) # т.е. происходит запуск на двух узлах с использованием 20 ядер на каждом. # количество mpiprocs=2*PPN, потому что gamess использует 2 mpi процесса на каждом ядре. rungms SubPc.inp 01 40 20 >& run.log
Дополнительно¶
Значение переменной для задания внешнего базисного набора атомных орбиталей установлено как:
setenv EXTBAS $HOME/EXTBAS.txtДля его использования достаточно разместить файл
EXTBAS.txt в своей домашней директории.
BioBuilds¶
BioBuilds — коллекция инструментов биоинформатики с открытым исходным кодом для работы в области геномики.
Установлена сборка 2017.11. Среди множества пакетов коллекции доступны:
| Trinity | 2.2.0 |
| Samtools | 1.6.0 |
| Bowtie | 1.1.2 |
| NCBI BLAST+ | 2.6.0 |
| R | 3.4.2 |
Для использования на кластере необходимо подключить модуль:
module load biobuilds/2017.11
ModEM¶
ModEM — свободно доступная программа для 3D инверсии MT данных.
Установлена версия от 2016.05.
Для использования на кластере необходимо подключить модуль:
module load modem/2016.05Эта команда добавит нужный путь к исполняемым файлам:
Mod2DMT Mod2DMT_seq Mod3DMT Mod3DMT_seq
Справочная информация¶
Описание основных команд при работе в ОС Linux — http://wwwinfo.jinr.ru/unixinfo/pc/lin_os.html
Документация к системе диспетчеризации заданий PBS Pro — http://www.pbsworks.com/pdfs/PBSUserGuide14.2.pdf
{kind=link}